





熱エネルギー工学研究室では、熱エネルギーの輸送・貯蔵・変換の研究を、分子スケールから連続体までの幅広いスケールで行っています。熱科学、流体力学、固体物理学、表面化学の融合による基礎研究から新しい現象を探求し、熱電変換を利用した発電、電子デバイスや電気自動車の熱マネージメント、機器や建築の放射冷却のような様々な応用に資する材料・デバイス・システムを開発しています。これによって、熱エネルギーの効率的な活用や省エネルギー技術の革新を推進し、持続可能な社会への貢献を目指しています。
Mission
持続的社会の実現のために、
熱エネルギーを有効利用する。
Research
-
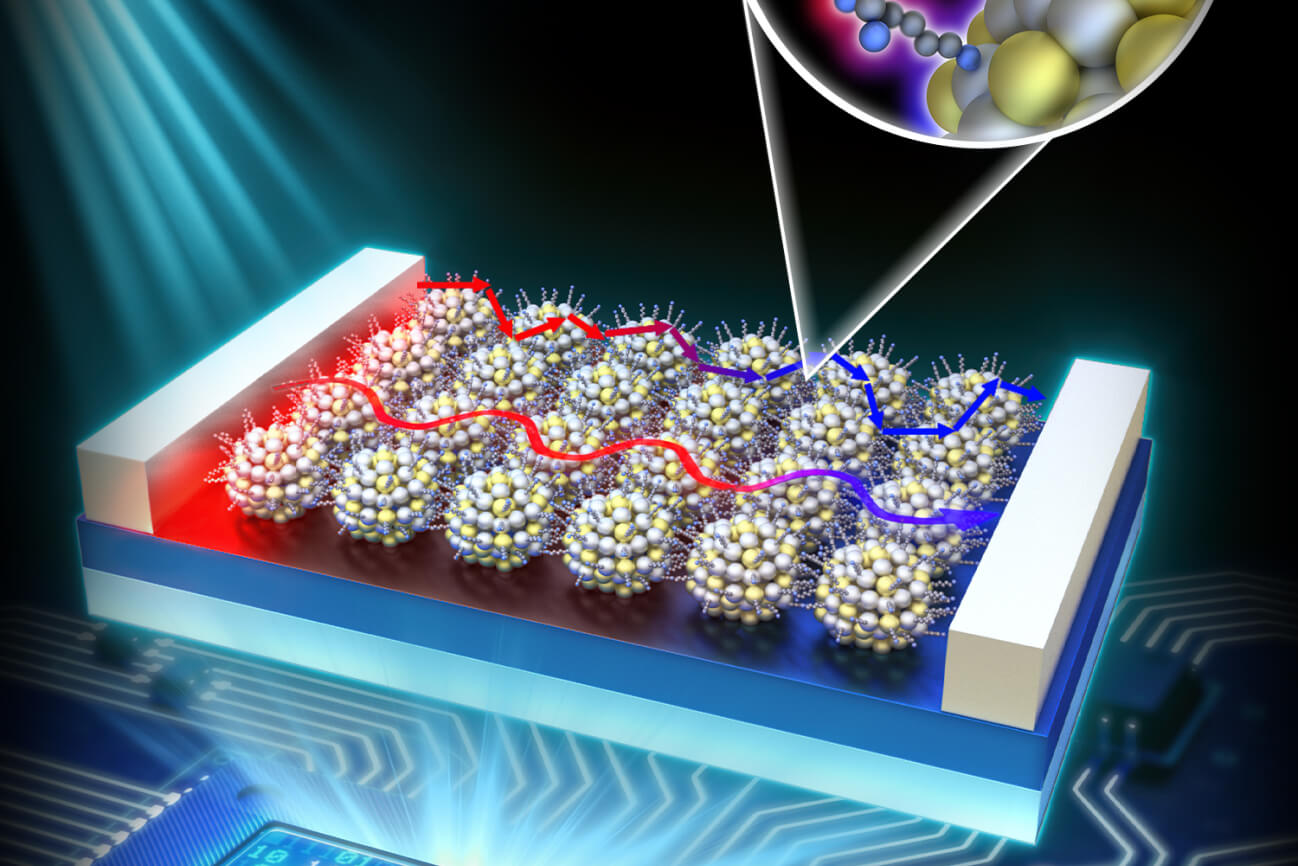
フォノンエンジニアリング
熱マネージメント/フォノン輸送/電子/マグノン/ナノ構造/熱伝導計測技術/マルチスケールシミュレーション/第一原理計算
-

マテリアルズ・
インフォマティクス材料探索/構造探索/巨大計算空間/量子コンピューター/量子アニーラー/超熱物性材料開拓/ロボティクス/機械学習/ラボオートメーション/実験自動化
-

熱電変換材料とデバイス
環境発電/IoT/熱電特性/ゼーベック効果/シリコン/自然冷却
-

動的濡れ現象の制御
固-液接触線/接触線抵抗/接触角ヒステリシス/高速濡れ/動的濡れ/表面微細構造/表面濡れ性制御/相変化/熱交換/熱伝達促進
-
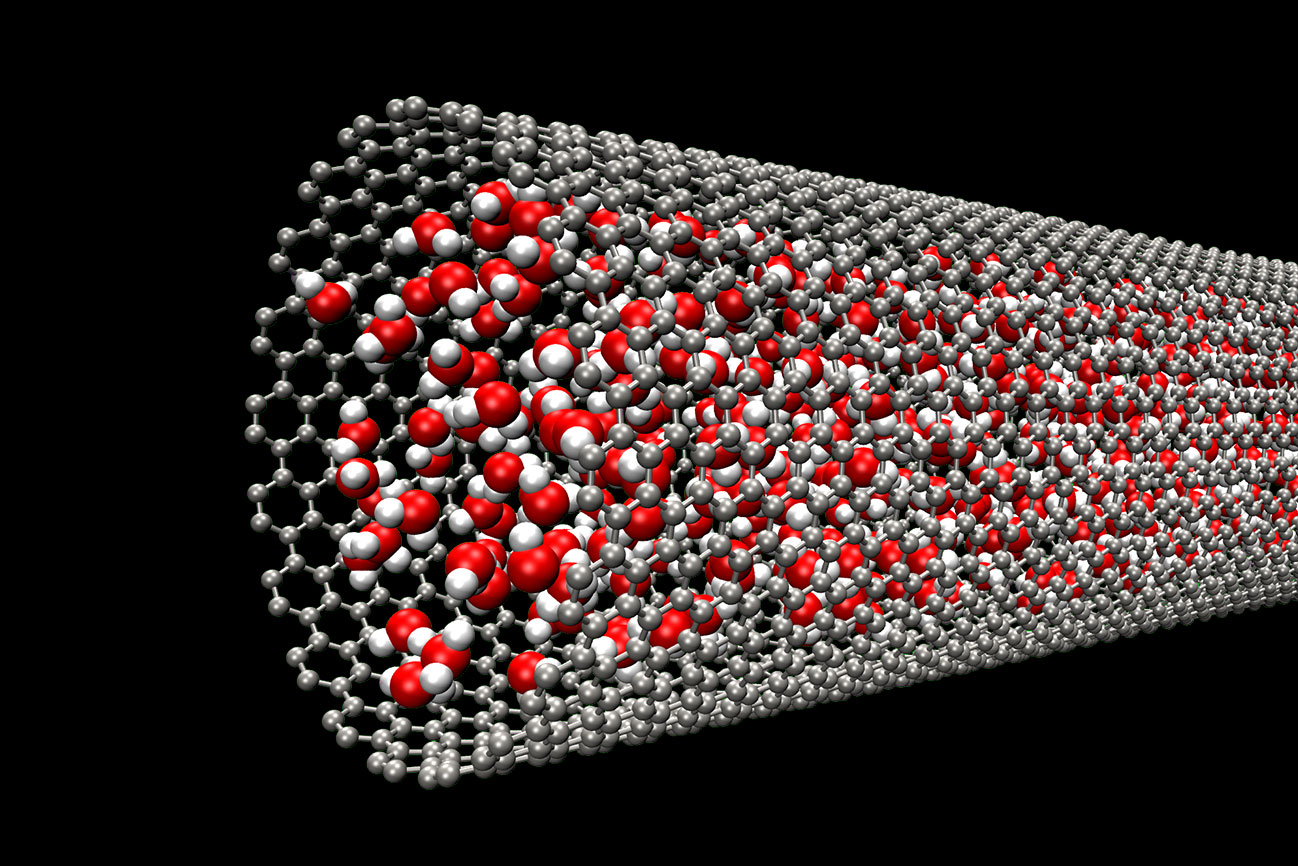
ナノ空間の熱流体力学
ナノ材料(カーボンナノチューブ、グラフェン、グラファイト、hBNなど)/インターカレーション/超イオン伝導体/熱輸送計算
-
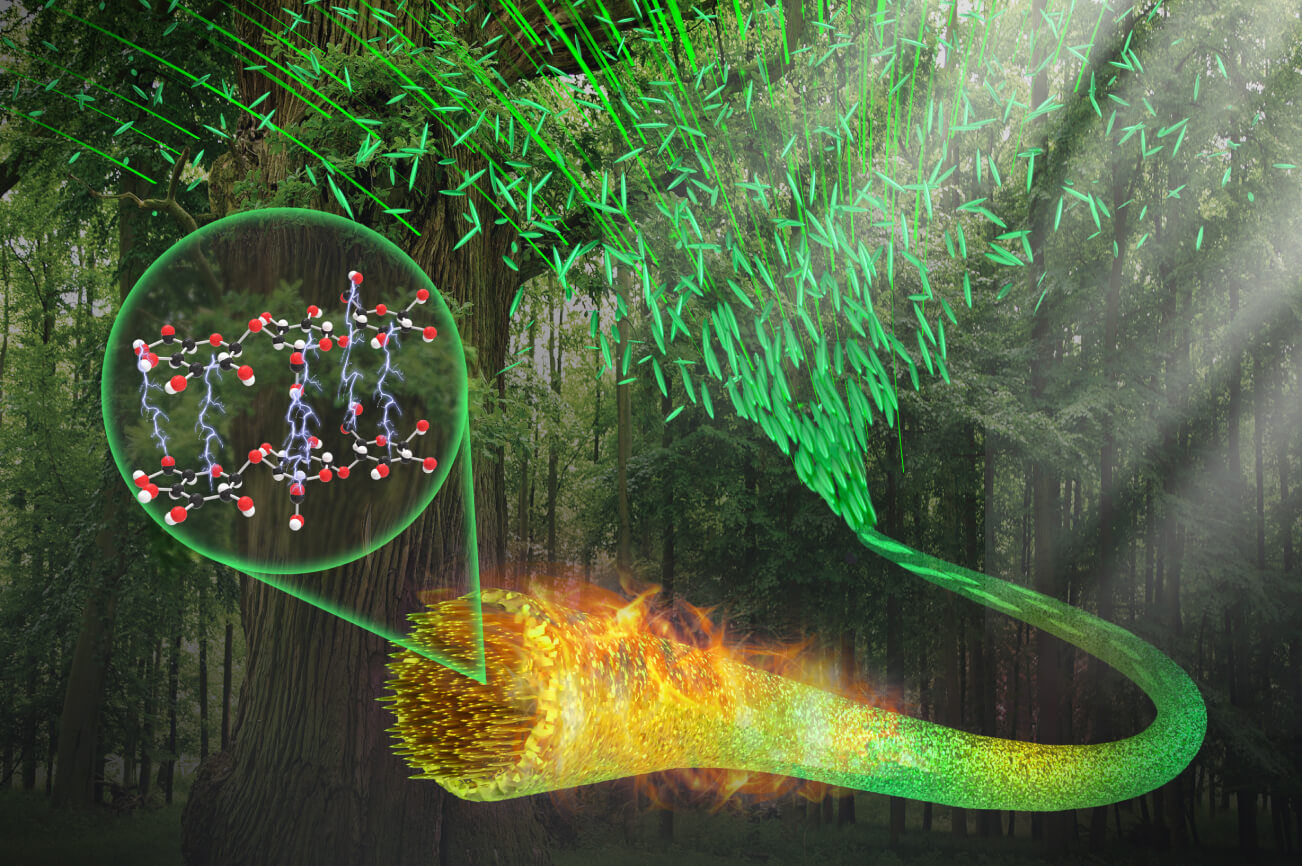
ナノセルロース材料の機能化
持続可能社会/セルロースナノファイバー(CNF)/階層構造/CNF配向制御/熱・電気的機能性新規材料開発













